|
January 2024
Billing chart: Blue Cross highlights medical, benefit policy changes
You’ll find the latest information about procedure codes and Blue Cross Blue Shield of Michigan billing guidelines in the following chart.
This billing chart is organized numerically by procedure code. Newly approved procedures will appear under the New Payable Procedures heading. Procedures for which we have changed a billing guideline or added a new payable group will appear under Updates to Payable Procedures. Procedures for which we are clarifying our guidelines will appear under Policy Clarifications. New procedures that are not covered will appear under Experimental Procedures.
We'll publish information about new Blue Cross groups or changes to group benefits under the Group Benefit Changes heading.
For more detailed descriptions of the Blue Cross' policies for these procedures, check under the Commercial Policy tab in Benefit Explainer on Availity®. To access this online information:
1. Log in to availity.com.
2 .Click on Payer Spaces on the Availity menu bar.
3. Click on the BCBSM and BCN logo.
4. Click on Benefit Explainer on the Applications tab.
5. Click on the Commercial Policy tab.
6. Click on Topic.
7. Under Topic Criteria, click on the circle for Unique Identifier and click the drop-down arrow next to Choose Identifier Type, then click on HCPCS Code.
8. Enter the procedure code.
9. Click on Finish.
10. Click on Search.
| Code* |
BCBSM changes to:
Basic Benefit and Medical Policy, Group
Variations Payment Policy, Guidelines
|
| NEW PAYABLE PROCEDURES |
93702 |
Basic benefit and medical policy
Bioimpedance for lymphedema
Devices using bioimpedance (bioelectrical impedance
spectroscopy) in the diagnosis, surveillance of cancer-
related extremity asymptomatic sub-clinical lymphedema is considered established when criteria is met. This policy was effective Sept. 1, 2023.
Payment policy:
Modifiers 26 and TC don’t apply to this procedure.
Inclusions:
Individuals meeting one of the following criteria:
- Regional (axillary or inguinal) lymph node dissection or sentinel lymph node biopsy with > 6 nodes removed
- Regional node irradiation
- Taxane-based chemotherapy
Note: L-Dex measurements should be performed as (1) A baseline before surgery/treatment. (2) Quarterly for the first year post-operatively. (3) Everly six months on years 2 and 3 post-operatively. (4) Yearly for years 4 and 5 post-operatively.
Exclusions:
|
| POLICY CLARIFICATIONS |
00170,** 41899**
**Unlisted procedure code |
Basic benefit and medical policy
Dental anesthesia
The safety and efficacy of general anesthesia and intravenous sedation for specified dental procedures have been established. They are useful therapeutic options for individuals meeting the appropriate patient selection criteria.
The inclusionary and exclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
This policy addresses general anesthesia provided for rendering dental services. General anesthesia services for rendering a dental procedure can be eligible for separate reimbursement if the anesthesia is rendered by a provider other than the dental services provider (such as an anesthesiologist or certified registered nurse anesthetist, or CRNA). All facility charges incurred in association with the anesthesia charges are covered under the medical/surgical benefit if any one of the following criteria is met:
- For children under age 7 (i.e., through the end of the 6th year)
- For older patients (age 7 and older), consider the extent of the procedures required. At a minimum, the individual should require one of the following:
- A total of six or more teeth to be extracted.
- Procedures that must be performed in two or more quadrants of the mouth on the same date of service.
In addition, for patients age 7 and older, documentation of one of the following conditions must exist:
- A concurrent hazardous medical or behavioral condition that creates a documented medical necessity for performing the procedure in an accredited facility using general anesthesia or sedation. These conditions may include, but are not limited to, labile hypertension, significant cardiac arrhythmias (more than five premature ventricular contractions per minute on EKG), Down’s syndrome, cerebral palsy or spasticity, morbid obesity, autism, movement disorders, chronic respiratory disease, hemophilia and other bleeding disorders, uncontrolled diabetes, etc. Note: A history of chronic diabetes mellitus isn’t considered a concurrent hazardous medical condition under the above criteria.
- A statement from the member’s primary physician supporting that the member has medical conditions too serious to undergo dental treatment in the dental office setting.
- Significant cellulitis or swelling and associated trismus (a sustained spasm of the jaw muscles, characteristic of the early stages of tetanus) that doesn’t allow the use of local anesthesia.
- Extensive orofacial or dental trauma for which treatment under local anesthesia would be ineffective or compromised.
- Extremely uncooperative, fearful, unmanageable or anxious, as determined by their primary care provider, or uncommunicative members with dental needs of such magnitude that treatment shouldn’t be postponed or deferred and for whom lack of treatment can be expected to result in dental or oral pain, infection, loss of teeth or other increased oral or dental morbidity.
- Allergy or sensitivity to local anesthesia
Exclusions:
- All other anesthesia modalities used to perform dental services in the dental provider’s office.
|
11920, 11921, 11922, 17380, 17999,** 19318, 19325, 19350, 21120, 21121, 21122, 21123, 21125, 21127, 21137, 21138, 21139, 21209, 30400, 30410, 30420, 31599,** 31899,** 54520, 55970, 55980, 56805, 57291, 57292, 57335, 58150, 58152, 58180, 58260, 58262, 58275, 58291, 58541, 58542, 58543, 58544, 58550, 58552, 58553, 58554
Experimental
11950, 11951, 11952, 11954, 15769, 15771, 15772, 15773, 15774, 15820, 15821, 15822, 15823, 15824, 15825, 15826, 15828, 15830, 15832, 15833, 15834, 15835, 15836, 15837, 15838, 15839, 15876, 15877, 15878, 15879, 19316, 21208, 30430, 30435, 30450, 69300, Q2026, Q2028
**Unlisted codes |
Basic benefit and medical policy
Lung and lobar lung transplants
Gender affirming services
The safety and effectiveness of select medical and surgical treatments for gender dysphoria have been established. The established treatments for gender dysphoria include:
- Puberty suppression in adolescents
- Hormone therapy (for masculinization or feminization) for adolescents and adults who meet criteria.
- Medically necessary gender-affirming surgery:
- Genitalia reconstruction
- Mastectomy — defined in this policy as surgical removal of the breast
- Augmentation mammoplasty (implants)
- Thyroid reduction chondroplasty (tracheal shave)
- Facial feminization
- Facial masculinization
Note: Surgery can’t be used to reverse effects of hormone therapy used to affirm an individual’s gender identity/expression.
To ensure appropriate preventive medical care, any anatomical structure present that warrants screening should continue to be screened, regardless of gender identity. Examples include:
- Breast cancer screening in transgender and gender-diverse people with breasts formed during natal puberty who haven’t undergone gender-affirming chest surgery and for transgender and gender-diverse people who have received estrogens, taking into account the length of time of hormone use, dosing, current age and age at which the hormones were initiated
- Prostate cancer screening for transgender and gender diverse people who have retained their prostate
- Cervical screening for transgender and gender-diverse people who currently have or previously had a cervix following local guidelines for individuals assigned female at birth.
- Obstetric services for transgender and gender diverse people when they are pregnant.
The medical policy statement, and inclusionary and exclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
While the following procedures are considered established for this policy, the specific coverage for each member is based on the benefit, which is defined by the group or employer.
Assessment, diagnosis and treatment should be provided through a multidisciplinary gender services clinic or program affiliated with a major medical center. If this level of service is unavailable, there should be documentation that reflects a coordinated approach to care by specialists involved (mental health specialists, physicians, surgeons, etc.).
Puberty suppression**
Health care professionals taking care of transgender and gender diverse adolescents with gender dysphoria should involve relevant disciplines, including mental health and medical professionals, to reach a decision about whether puberty suppression or hormone initiation for these adolescents are appropriate and remain indicated throughout the course of treatment until the transition is made to adult care. Puberty suppression hormones for adolescents may be indicated for members who meet all the following inclusionary criteria:
Inclusions:
- Meets diagnostic criteria for gender dysphoria (see description and background above for diagnostic criteria).
- Gender dysphoria is marked and sustained.
- Demonstrates emotional and cognitive maturity required to provide informed consent for the treatment and, particularly when the adolescent hasn’t reached the age of medical consent, the parents, guardians or other legally authorized caretakers have consented to the treatment and are involved in supporting the adolescent throughout the treatment process.
- Mental health concerns (if any) that may interfere with diagnostic clarity, capacity to consent and gender-affirming medical treatments have been addressed — sufficiently so that gender-affirming medical treatment can be provided optimally.
- Onset of puberty to at least Tanner stage 2 has been reached.
- Contraindications to therapy are absent in the judgment of the managing physician.
**Medications for puberty suppression may be managed under the member’s pharmacy benefit.
Hormone therapy**
Hormone therapy may be indicated for members who meet all the following inclusionary criteria.
Inclusions for adolescents, defined as from the start of puberty until the legal age of majority (18 years of age):
- Meets diagnostic criteria for gender dysphoria (see description and background above for diagnostic criteria).
- Gender dysphoria is marked and sustained.
- Other possible causes of apparent gender incongruence have been identified and excluded.
- Demonstrates emotional and cognitive maturity required to provide informed consent for the treatment and, particularly when the adolescent hasn’t reached the age of medical consent, the parents, guardians or other legally authorized caretakers have consented to the treatment and are involved in supporting the adolescent throughout the treatment process.
- Mental health concerns (if any) that may interfere with diagnostic clarity, capacity to consent and gender-affirming medical treatments have been addressed — sufficiently so that gender-affirming medical treatment can be provided optimally.
- Onset of puberty to at least Tanner stage 2 has been reached.
- Contraindications to therapy are absent in the judgment of the managing physician.
- Mental health and physical conditions that could negatively impact the outcome of gender-affirming medical treatments have been assessed, with the risks and benefits discussed, before a decision is made regarding treatment.
- One letter of assessment as indicated below.***
- Medications will be prescribed by or in consultation with a pediatric endocrinologist who has collaborated on care with a mental health care provider.
Inclusions for adults:
- 18 years of age or older (age of majority).
- Meets diagnostic criteria for gender dysphoria (see description and background above for diagnostic criteria).
- Gender dysphoria is marked and sustained.
- Other possible causes of apparent gender incongruence have been identified and excluded.
- Demonstrates capacity to make a fully informed decision and to consent for gender-affirming hormone treatment.
- Contraindications to therapy are absent in the judgment of the managing physician.
- Mental health and physical conditions that could negatively impact the outcome of gender-affirming medical treatments have been assessed, with the risks and benefits discussed, before a decision is made regarding treatment.
**Medications for hormone therapy may be managed under the member’s pharmacy benefit.
Hair removal
- If gender-affirming surgery is approved for a transgender or gender diverse person, hair removal (e.g., electrolysis, laser hair removal) may be considered established following medical review only for the types of tissue described below:
- When the scrotal and surrounding tissues are used in the surgical construction of the vagina.
- When free flap or other donor tissues are used for phalloplasty performed in conjunction with vaginectomy and full-length urethroplasty.
Gender-affirming surgery
Inclusions:
Gender-affirming surgery may be indicated for members who meet all the following inclusionary criteria:
- Chest surgery**
- Mastectomy (surgical removal of the breast) is considered reconstructive when all the following criteria has been met:
- The individual is at least 18 years of age.
- The individual has been diagnosed with gender dysphoria (see the description and background section for diagnostic criteria).
- Gender dysphoria is marked and sustained.
- The individual has the capacity to make fully informed decisions and consent for treatment.
- One letter of assessment as indicated below.***
- Other possible causes of apparent gender incongruence have been identified and excluded.
- Mental health and physical conditions that could negatively impact the outcome of gender-affirming medical treatments have been assessed, with the risks and benefits discussed, before a decision is made regarding treatment.
- Hormone therapy prior to mastectomy isn’t required as the aim of hormone therapy prior to facial surgery or gonadectomy is primarily to introduce a period of reversible testosterone or estrogen suppression before the individual undergoes irreversible surgical intervention.
- Living in a gender role congruent with gender identity for 12 continuous months isn’t required prior to a mastectomy.
- Pre-operative and post-operative care that addresses both surgical results and possible behavioral health impacts is highly recommended.
- Nipple reconstruction, including tattooing, following a gender-affirming mastectomy that meets the reconstructive criteria above is considered reconstructive.
- Breast augmentation is considered reconstructive when all the following criteria has been met:
- The individual is at least 18 years of age.
- The individual has been diagnosed with gender dysphoria (see the description and background section for diagnostic criteria).
- Gender dysphoria is marked and sustained.
- The individual has the capacity to make fully informed decisions and consent for treatment.
- One letter of assessment as indicated below.***
- Other possible causes of apparent gender incongruence have been identified and excluded.
- Mental health and physical conditions that could negatively impact the outcome of gender-affirming medical treatments have been assessed, with the risks and benefits discussed, before a decision is made regarding treatment.
- The individual is stable on their gender-affirming hormonal treatment regime for at least 12 months, unless a rationale is provided by the health care practitioner that indicates that hormone treatment is either contraindicated or not necessary for the individual’s clinical situation.
- Living in a gender role congruent with gender identity for 12 continuous months isn’t required prior to breast augmentation.
- Existing chest appearance demonstrates significant variation from expected appearance for the individual’s gender identity.
- Pre-operative and post-operative care that addresses both surgical results and possible behavioral health impacts is highly recommended.
**The procedures needed to reconstruct a feminine/masculine appearance can only be performed once per lifetime.
- Facial surgery**† is considered reconstructive when all the following criteria has been met:
- The individual is at least 18 years of age.
- The individual has been diagnosed with gender dysphoria (see the description and background section for diagnostic criteria).
- Gender dysphoria is marked and sustained.
- The individual has the capacity to make fully informed decisions and consent for treatment.
- Other possible causes of apparent gender incongruence have been identified and excluded.
- One letter of assessment as indicated below.***
- Mental health and physical conditions that could negatively impact the outcome of gender-affirming medical treatments have been assessed, with the risks and benefits discussed, before a decision is made regarding treatment.
- The individual is stable on their gender-affirming hormonal treatment regime for at least 12 months, unless a rationale is provided by the health care practitioner that indicates that hormone treatment is either contraindicated or not necessary for the individual’s clinical situation.
- The gender identity should be present for at least 12 months.
- The member has a consistent stable gender identity that is well documented by their treating providers and, when possible, lives as their affirmed gender in places where it is safe to do so.
- Existing facial appearance demonstrates significant variation from expected appearance for the individuals gender identity (photographic evidence must be maintained as part of the medical record).
- The procedure directly addresses variation from expected appearance for the experienced gender. (Note: Each procedure requested should be considered separately as some procedures may be cosmetic and others may be reconstructive.)
- Pre-operative and post-operative care that addresses both surgical results and possible behavioral health impacts is highly recommended.
**The procedures needed to reconstruct a feminine/masculine appearance can only be performed once per lifetime.
†Procedures included in this group that aren’t considered cosmetic when used to treat gender dysphoria are:
- Thyroid reduction chondroplasty (tracheal shave)
- Genioplasty (repositioning or reshaping of the chin) Mandible augmentation (jawline contouring/reconstruction)
- Facial bone reduction
- Forehead reduction/contouring
- Rhinoplasty (reshaping/contouring of the nose).
- Genital surgery is considered medically necessary when all the following criteria have been met:
- The individual is at least 18 years of age.
- The individual has been diagnosed with gender dysphoria (see the description and background section for diagnostic criteria).
- Gender dysphoria is marked and sustained.
- The individual has the capacity to make fully informed decisions and consent for treatment.
- Other possible causes of apparent gender incongruence have been identified and excluded.
- One letter of assessment as indicated below.***
- Mental health and physical conditions that could negatively impact the outcome of gender-affirming medical treatments have been assessed, with the risks and benefits discussed, before a decision is made regarding treatment.
- The individual is stable on their gender-affirming hormonal treatment regime for at least 12 months, unless a rationale is provided by the health care practitioner that indicates that hormone treatment is either contraindicated or not necessary for the individual’s clinical situation.
- The gender identity should be present for at least 12 months.
- The member has a consistent stable gender identity that is well documented by their treating providers and, when possible, lives as their affirmed gender in places where it is safe to do so.
- Pre-operative and post-operative care that addresses both surgical results and possible behavioral health impacts is highly recommended.
These criteria don’t apply to patients who are having these surgical procedures for medical indications other than gender dysphoria.
***Letter requirements:
- Required for hormone therapy for adolescents
- Required for facial, pelvic, gonadal or genital surgery for adults
Adolescents
One letter of assessment from the multidisciplinary team (or in situations where a multidisciplinary team is not available, a professional from one of the multiple disciplines who are experts in transgender health and in the management of the care required for transgender and gender diverse adolescents who is taking care of the individual) is required for adolescents receiving gender-affirming medical treatment. This letter needs to reflect the assessment and opinion from the team, which involves both medical health care practitioners and mental health professionals, and support that the individual meets the criteria for gender dysphoria. Health care professionals working with gender-diverse adolescents should undertake a comprehensive biopsychosocial assessment of adolescents who present with gender identity-related concerns and seek medical related care, and accomplish this in a collaborative and supportive manner. The detailed assessment must have been performed within 12 months of the first requested treatment.
The health care professional assessing and working with the adolescent should meet all the following criteria:
- Are licensed by their professional body and hold a postgraduate degree or its equivalent in a clinical field related to transgender health granted by a nationally accredited institution.
- Receive theoretical and evidenced-based training and develop expertise in general child, adolescent and family mental health across the developmental spectrum.
- Receive training and have expertise in gender identity development, gender diversity in children and adolescents, have the ability to assess capacity to assent/consent and possess general knowledge of gender diversity across the lifespan.
- Continue engaging in professional development in all areas relevant to gender-diverse children, adolescents and families.
- Receive training and develop expertise in autism spectrum disorders and other neurodevelopmental presentations or collaborate with a developmental disability expert when working with autistic/neurodivergent gender diverse adolescents.
Adults
One letter of assessment from a health care professional who has competencies in the assessment of transgender and gender diverse people, documenting that the individual meets the criteria for gender dysphoria, is required for transgender and gender-diverse adults for gender-affirming surgical treatments. The detailed assessment must have been performed within 12 months of the initial requested procedure.
The health care professional should meet all the following criteria:
- Are licensed by their professional body and hold, at a minimum, a master’s degree or equivalent training in a clinical field related to transgender health or equivalent further clinical training in this area that’s granted by a nationally accredited institution
- Should be competent using the latest edition of the Diagnostic and Statistical Manual of Mental Disorders for diagnosis
- Are able to identify co-existing mental health or other psychosocial concerns and distinguish these from gender dysphoria, incongruence and diversity
- Are able to assess capacity to consent for treatment
- Have experience or be qualified to assess clinical aspects of gender dysphoria, incongruence and diversity
- Undergo continuing education in health care relating to gender dysphoria, incongruence and diversity
Exclusions:
- Gender-affirming services aren’t covered if contract or certificate language contains specific exclusion of these services.
- Reversal of physical or functional outcomes due to gender-affirming services.
- Reversal of surgical procedures performed for gender affirming services.
- All procedures that are primarily cosmetic and not reconstructive or not medically necessary including, but not limited to:
- Abdominoplasty
- Blepharoplasty
- Brow lift
- Calf implants
- Cheek/malar implants
- Chin/nose implants
- Collagen injections
- Drugs for hair loss or growth
- Forehead lift
- Hair removal (for exception: see Inclusions, electrolysis)
- Hair transplantation
- Injectable dermal fillers (e.g., Sculptra, Radiesse)
- Lip reduction
- Liposuction
- Mastopexy
- Neck tightening
- Otoplasty
- Pectoral implants
- Removal of redundant skin
- Rhytidectomy
- Speech language therapy
- Non-covered services
|
17999,** 20999**
**Unlisted code |
Basic benefit and medical policy
Percutaneous ultrasonic ablation (Tenex Health TX)
The use of the Tenex Health TX® procedure is considered experimental when used to treat tendon pain, regardless of the anatomical location. There is insufficient published evidence to assess the safety, impact on health outcomes or patient management of the Tenex Health TX procedure for treatment of tendon pain, effective Jan. 1, 2024.
Inclusionary and exclusionary guidelines:
Not applicable |
19318 |
Basic benefit and medical policy
Breast reduction services
The safety and effectiveness of breast reduction have been established. It may be considered a useful therapeutic option (and not considered cosmetic) when one of the following is met:
- Individual selection guidelines in this policy are met.
- Performed in conjunction with medically necessary breast reconstruction for the purposes of attaining breast symmetry.**
**Refer to the medical policy “Reconstructive Breast Surgery/Management of Implants.”
The medical policy statement was updated, effective Jan. 1, 2024.
Inclusions:
Patient selection guidelines
Patients under the age of 18 years can’t give legal consent for surgery. The parent or legal guardian must support and authorize a reduction mammaplasty (breast reduction).
Emancipated minors may be extended individual consideration.
Inclusions:
Must meet A, or must meet both B and C
- Must meet both 1 and 2:
- Patient’s breasts are fully grown (i.e., breast size stable for approximately one year)
- Removal of more than 500 grams of tissue from each breast
- One of the following (1, 2 or 3) must be met:
- Pain (both of the following)
- Documented pain in the neck and/or shoulders or postural backache which must be of long-standing duration.
- Failure of conservative therapy (e.g., an appropriate support bra, exercises, heat/cold treatments, non-steroidal anti-inflammatory agents or muscle relaxants).
- Shoulder grooving
- Recurrent intertrigo between the breasts and the chest wall
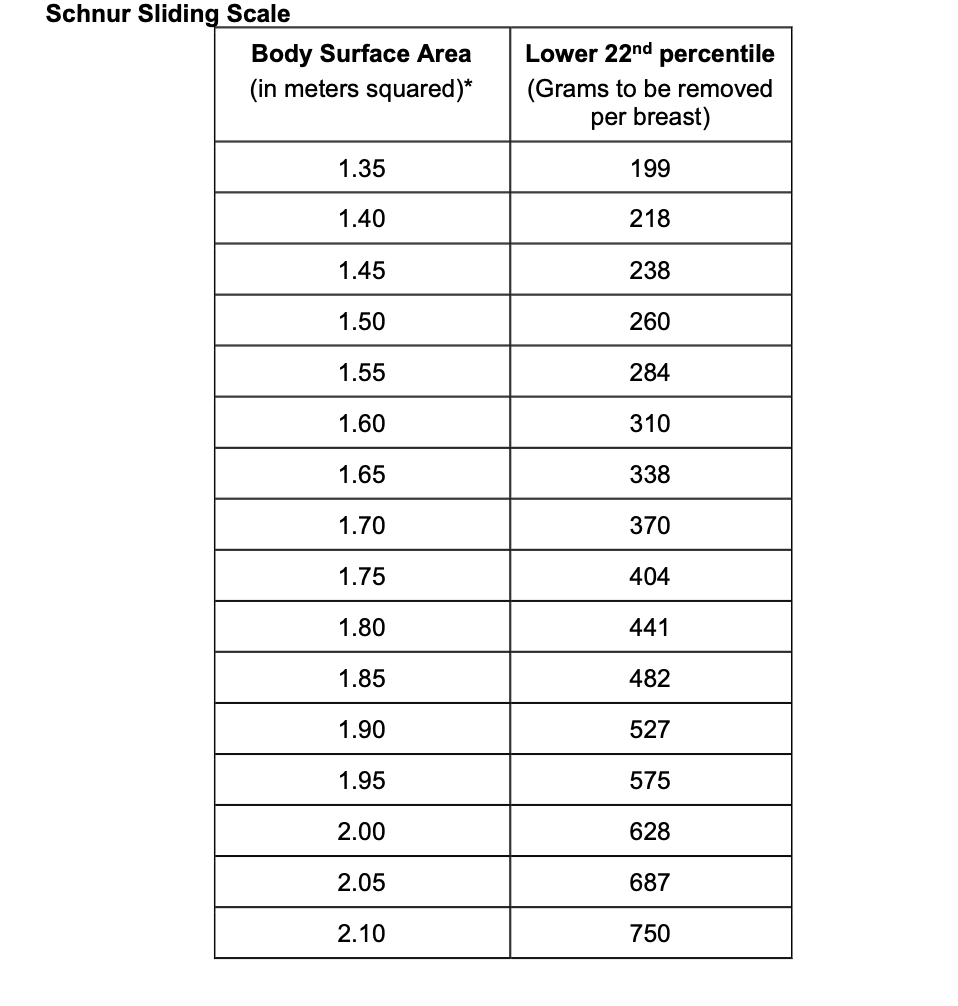
- Both of the following criteria must be met:
- Breasts are fully grown (i.e., breast size stable for approximately one year).
- The amount of tissue to be removed from each breast must be greater than or equal to the 22nd percentile on the Schnur Scale.**
**If one breast meets the tissue amount based on the Schnur Scale (even if the other breast doesn’t), this criterion is met.
If one breast meets the Schnur scale criteria, and all other criteria for breast reduction are met, the breast tissue may be removed from the other breast to achieve symmetry.
The Schnur Sliding Scale (see below) is used by physicians to evaluate individuals being considered for breast reduction surgery.
Body surface area, along with average weight of breast tissue removed is incorporated into the chart. If the individual's body surface area and weight of breast tissue removed fall below the 22nd percentile, then the surgery isn’t medically necessary. If the individual's body surface area and weight of breast tissue removed is above the 22nd percentile, then the surgery is considered medically necessary if other applicable criteria are met.
Calculation of body surface area
Body surface area = the square root of height (cm) times weight (kg) divided by 3600.
- To convert pounds to kilograms, multiply pounds by 0.45.
- To convert inches to meters, multiply inches by .0254.
To calculate body surface area, click here.
Exclusions:
Breast reduction isn’t covered for either of the following indications because it’s considered cosmetic in nature and not medically necessary:
- Surgery is being performed to treat psychological symptomatology or psychosocial complaints, in the absence of significant physical, objective signs.
- Surgery is being performed for the sole purpose of improving appearance.
|
20552, 20553, 20605, 20606, 21010,
21050, 21060, 21070, 21073, 21085,
21116, 21240, 21242, 21243, 21480,
21485, 21490, 29800, 29804, 70328,
70330, 70332, 70336, 70350, 70355,
70486, 70487, 70488, 97010, 97024
Experimental/not covered:
21089, 21299, 64615, E1399, J0585,
J7321, J7323, J7324, J7325, J7326 |
Basic benefit and medical policy
TMJ disorders
Certain tests, non-surgical and surgical procedures are considered safe and effective for the diagnosis and therapeutic treatment of temporomandibular joint, known as TMJ, disorders. They may be considered useful therapeutic options when indicated. Exclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
The following diagnostic procedures when used to diagnose TMJ dysfunction:
- Diagnostic X-ray, tomograms and arthrograms
- Medical grade CT scan or MRI (generally CT scans and MRIs are reserved for presurgical evaluations)
- Cephalograms (X-rays of jaws and skull)
- Pantograms (panoramic X-rays of maxilla and mandible)
The following non-surgical treatments for the treatment of TMJ dysfunction:
- Intraoral removable prosthetic devices or appliances (encompassing fabrication, insertion, adjustment) of any and all devices or appliances constructed (excludes dental devices — see below)
- Pharmacologic treatment (e.g., anti-inflammatory, muscle relaxing and analgesic medications)
- Trigger point therapy with anesthetic or corticosteroid for the treatment of myofascial pain syndrome, are limited to no more than four injections in a 12-month period, when all the following are met:
- There is a regional pain complaint in the expected distribution of referral pain from a trigger point.
- There is spot tenderness in a palpable taut band in a muscle.
- There is restricted range of motion.
- Conservative therapy (e.g., physical therapy, active exercises, ultrasound, heating or cooling, massage, activity modification, or pharmacotherapy) doesn’t result in adequate symptom relief within two to three weeks, or isn’t feasible.
- Trigger point injections are provided as a component of a comprehensive therapy program.
The following surgical procedures for the treatment of TMJ dysfunction:
- Arthrocentesis with or without ultrasound guidance
- Manipulation for reduction or dislocation of the TMJ
- Arthroscopic surgery in patients that objectively demonstrate (by physical examination or imaging) internal derangements (displaced discs) or degenerative joint disease who have failed conservative treatment.
- Open surgical procedures (when TMJ dysfunction results from congenital anomalies, trauma or disease in individuals who have failed conservative treatment) including, but not limited to, arthroplasties, condylectomies, condylotomies, meniscus or disc plication and disc removal.
Note: Dental restorations for reconstruction of tooth form and function that are a result of TMJ dysfunction or bruxism are considered a dental service and aren’t a covered medical-surgical benefit unless otherwise specified in the individual medical certificate.
Exclusions:
The following diagnostic procedures when used to diagnose bruxism** or TMJ dysfunction:
- Electromyography, including surface EMG
- Kinesiography
- Thermography
- Neuromuscular junction testing
- Somatosensory testing
- Transcranial or lateral skull X-rays
- Intra-oral tracing or gothic arch tracing (intended to demonstrate deviations in the positioning of the jaws that are associated with TMJ dysfunction)
- Muscle testing
- Standard dental radiographic procedures
- Range of motion measurements
- Computerized mandibular scan (this measures and records muscle activity related to movement and positioning of the mandible and is intended to detect deviations in occlusion and muscle spasms related to TMJ dysfunction)
- Ultrasound/sonogram (ultrasonic Doppler auscultation)
- Arthroscopy of the TMJ for purely diagnostic purposes
- Joint vibration analysis
- Cone beam computed tomography**
- Trigger point therapy for any indication not listed above
- Use of any medication not listed above (e.g., botulinum toxin, methylprednisolone)
- Image guidance of trigger point injections
The following non-surgical procedures for the treatment of TMJ dysfunction:
- Electrogalvanic stimulation
- Iontophoresis
- Biofeedback
- Ultrasound
- Devices promoted to maintain joint range of motion and to develop muscles involved in jaw function.
- Orthodontic services or treatment (e.g., dental appliance that is intended to treat malocclusion by tooth and support structure movement)
- Dental restorations, prosthesis, treatment and appliances**
- Transcutaneous electrical nerve stimulation
- Percutaneous electrical nerve stimulation
- Acupuncture
- Platelet concentrates
- Dextrose prolotherapy
- Botulinum toxin A
**Intra-oral reversible orthotic device (also known as occlusal orthotic, occlusal guard or bite splint), including fabrication, insertion and adjustment of all devices fabricated, cone beam tomography and bruxism treatment are exclusions in most cases. Refer to current certificate of coverage for details.
|
29800, 29805, 29830, 29840, 29860, 29870, 29900, 29999**
**Unlisted code |
Basic benefit and medical policy In-office arthroscopy
In-office needle arthroscopy using the mi-eye 2™, mi-eye 3 needlescope™ with cannula and VisionScope® is experimental. Its use hasn’t been scientifically demonstrated to improve patient clinical outcomes.
The medical policy statement has been updated, effective Jan. 1, 2024.
Inclusionary and exclusionary guidelines:
Not applicable |
33215, 33216, 33217, 33218, 33220, 33223, 33230, 33231, 33240, 33241, 33243, 33244, 33249, 33262, 33263, 33264, 33270, 33271, 33272, 33273, 93260, 93261, 93282, 93283, 93284, 93287, 93289, 93295, 93296, 93297
Experimental
0571T, 0572T, 0573T, 0574T, 0575T, 0576T, 0577T, 0578T, 0614T |
Basic benefit and medical policy
Implantable cardioverter defibrillator
The safety and effectiveness of a transvenous automatic implantable cardioverter defibrillator, or ICD, have been established. It may be considered a useful therapeutic option for patients who meet selection criteria.
The safety and effectiveness of a subcutaneous automatic implantable cardioverter defibrillator have been established. It may be considered a useful therapeutic option for patients who meet selection criteria.
The use of a substernal implantable cardioverter defibrillator is considered experimental. The safety and effectiveness on clinical outcomes haven’t been definitively demonstrated.
The medical policy statement, as well as inclusionary and exclusionary criteria, have been updated, effective Jan. 1, 2024.
Inclusionary and exclusionary guidelines:
Transvenous automatic implantable cardioverter defibrillators
I. Adults
The use of the automatic implantable cardioverter defibrillator, or AICD, may be considered established in individuals who meet the following criteria:
Primary prevention
Inclusions:
- Ischemic cardiomyopathy with New York Heart Association, or NYHA, functional Class II or Class III symptoms, a history of myocardial infarction at least 40 days before ICD treatment and left ventricular ejection fraction of 35% or less
- Ischemic cardiomyopathy with NYHA functional Class I symptoms, a history of myocardial infarction at least 40 days before ICD treatment and left ventricular ejection fraction of 30% or less
- Nonischemic dilated cardiomyopathy and left ventricular ejection fraction of 35% or less after reversible causes have been excluded and the response to optimal medical therapy has been adequately determined
- Hypertrophic cardiomyopathy, or HCM, with one or more major risk factors for sudden cardiac death (history of premature HCM-related sudden death in one or more first-degree relatives younger than 50 years; left ventricular hypertrophy greater than 30 mm; one or more runs of nonsustained ventricular tachycardia at heart rates of 120 beats per minute or greater on 24-hour Holter monitoring; prior unexplained syncope inconsistent with neurocardiogenic origin) and judged to be at high risk for sudden cardiac death by a physician experienced in the care of individuals with HCM
- Diagnosis of any one of the following cardiac ion channelopathies** and considered to be at high risk for sudden cardiac death.
- Congenital long QT syndrome
- Brugada syndrome
- Short QT syndrome
- Catecholaminerigic polymorphic ventricular tachycardia
- Diagnosis of cardiac sarcoid and considered to be at high risk for sudden cardiac death as indicated given one of the following below:
- Spontaneous sustained ventricular arrhythmias, including prior cardiac arrest if meaningful survival of greater than one year is expected
- Left ventricular ejection fraction, or LVEF, 35% or less, despite optimal medical therapy and a period of immunosuppression (if there is active inflammation), if meaningful survival of greater than one year is expected
- LVEF greater than 35% if meaningful survival of greater than one year is expected and one of the following:
- Syncope or near-syncope, felt to be arrhythmic in etiology
- Evidence of myocardial scar by cardiac MRI or positron emission tomographic scan
- Inducible sustained ventricular arrhythmias (>30 seconds of monomorphic VT or polymorphic VT) or clinically relevant VF
- An indication for permanent pacemaker implantation.
**Criteria for ICD implantation in individuals with cardiac ion channelopathies:
Individuals with cardiac ion channelopathies may have a history of a life-threatening clinical event associated with ventricular arrhythmic events, such as sustained ventricular tachyarrhythmia, after reversible causes, in which case they should be considered for ICD implantation for secondary prevention even if they don’t meet criteria for primary prevention.
Criteria for ICD placement in individuals with cardiac ion channelopathies derive from results of clinical input, a 2013 consensus statement from the HRS, European Heart Rhythm Association and the Asia-Pacific Heart Rhythm Society on the diagnosis and management of individuals with inherited primary arrhythmia syndromes, and a report from the HRS and EHRA's Second Consensus Conference on Brugada syndrome.
Indications for consideration for ICD placement for each cardiac ion channelopathy are as follows:
- Long QT syndrome, or LQTS:
- Individuals with a diagnosis of LQTS who are survivors of cardiac arrest
- Individuals with a diagnosis of LQTS who experience recurrent syncopal events while on β-blocker therapy
- Brugada syndrome, or BrS:
- Individuals with a diagnosis of BrS who are survivors of cardiac arrest
- Individuals with a diagnosis of BrS who have documented spontaneous sustained ventricular tachycardia with or without syncope
- Individuals with a spontaneous diagnostic type 1 electrocardiogram who have a history of syncope, seizure or nocturnal agonal respiration judged to be likely caused by ventricular arrhythmias (after noncardiac causes have been ruled out)
- Individuals with a diagnosis of BrS who develop ventricular fibrillation during programmed electrical stimulation
- Catecholaminergic polymorphic ventricular tachycardia, or CPVT:
- Individuals with a diagnosis of CPVT who are survivors of cardiac arrest
- Individuals with a diagnosis of CPVT who experience recurrent syncope or polymorphic/bidirectional VT despite optimal medical management or left cardiac sympathetic denervation
- Short QT syndrome, or SQTS:
- Individuals with a diagnosis of SQTS who are survivors of cardiac arrest
- Individuals with a diagnosis of SQTS who are symptomatic and have documented spontaneous VT with or without syncope
- Individuals with a diagnosis of SQTS who are asymptomatic or symptomatic and have a family history of sudden cardiac death
Note: For congenital LQTS, individuals may have one or more clinical or historical findings other than those outlined above that could, alone or in combination, put them at higher risk for sudden cardiac death. They can include individuals with a family history of sudden cardiac death due to LQTS, infants with a diagnosis of LQTS with functional 2:1 atrioventricular block, individuals with a diagnosis of LQTS in conjunction with a diagnosis of Jervell and Lange-Nielsen syndrome or Timothy syndrome, and individuals with a diagnosis of LQTS with profound QT prolongation (>550 ms). These factors should be evaluated on an individualized basis by a clinician with expertise in LQTS when considering the need for ICD placement.
Exclusions:
The use of the ICD for primary prevention is considered experimental in primary prevention individuals who have had one of the following:
- An acute myocardial infarction (less than 40 days before ICD treatment)
- New York Heart Association class IV congestive heart failure (unless patient is eligible to receive a combination cardiac resynchronization therapy ICD device)
- A cardiac revascularization procedure in past three months (coronary artery bypass graft or percutaneous transluminal coronary angioplasty) or are candidates for a cardiac revascularization procedure
- Noncardiac disease that would be associated with life expectancy less than one year
The use of the ICD for primary prevention is considered experimental for all other indications not meeting criteria.
Secondary prevention
Inclusions:
- Individuals with a history of a life-threatening clinical event associated with ventricular arrhythmic events, such as sustained ventricular tachyarrhythmia, after reversible causes (e.g., acute ischemia) have been excluded.
Exclusions:
- The use of the ICD for secondary prevention is considered experimental for all other indications not meeting criteria.
II. Pediatrics
Inclusions:
The use of the ICD or SCD may be considered established in pediatric individuals who meet any of the following criteria:
- Survivors of cardiac arrest, after reversible causes have been excluded
- Symptomatic, sustained ventricular tachycardia in association with congenital heart disease in individuals who have undergone hemodynamic and electrophysiologic evaluation
- Congenital heart disease with recurrent syncope of undetermined origin in the presence of either ventricular dysfunction or inducible ventricular arrhythmias
- Hypertrophic cardiomyopathy, or HCM, with one or more major risk factors for sudden cardiac death (history or premature HCM-related sudden death in one or more first-degree relatives younger than 50 years; massive left ventricular hypertrophy based on age-specific norms; prior unexplained syncope inconsistent with neurocardiogenic origin) and judged to be at high risk for sudden cardiac death by a physician experienced in the care of patients with HCM.
- Diagnosis of any one of the following cardiac ion channelopathies** (refer to Criteria for ICD implantation in individuals with cardiac ion channelopathies for adults above) and considered to be at high risk for sudden cardiac death:
- Congenital long QT syndrome
- Brugada syndrome
- Short QT syndrome
- Catecholaminergic polymorphic ventricular tachycardia
Exclusions:
The use of the transvenous ICD is considered experimental for all other indications in pediatric patients that don’t meet the criteria.
Subcutaneous automatic implantable cardioverter defibrillators
The use of a subcutaneous ICD may be considered established for adult or pediatric individuals who have an indication for transvenous ICD implantation for primary or secondary prevention for any of the above reasons and meet all the following criteria:
- Have a contraindication to a transvenous ICD due to one or more of the following: 1) lack of adequate vascular access; 2) compelling reason to preserve existing vascular access (e.g., need for chronic dialysis; younger individual with anticipated long-term need for ICD therapy); or 3) history of need for explantation of a transvenous ICD due to a complication with ongoing need for ICD therapy
- Have no indication for antibradycardia pacing
- Don’t have ventricular arrhythmias known or anticipated to respond to antitachycardia pacing
- A high risk for infection, e.g., immunocompromised patients or those with a history of a previous transvenous infection
- History of congenital heart disease with anatomic limitations for transvenous placement of the transvenous AICD
- History of need for explantation of a tranvenous ICD due to a complication, with an ongoing need for ICD therapy
Exclusions:
- The individual has a need for cardiac pacing.
- The use of subcutaneous ICD for all other indications that don’t meet the above criteria.
Substernal implantable cardioverter defibrillator
The substernal ICD is experimental for all indications. |
33945, 50360, 50365 |
Basic benefit and medical policy Heart-kidney transplant
The safety and effectiveness of a heart-kidney transplant have been established. It may be considered a useful therapeutic option for carefully selected patients with end-stage heart and kidney disease.
Inclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
Indications for combined heart-kidney transplant include, but aren’t limited to, progressive chronic heart and kidney disease unresponsive to other medical and surgical therapy. In general, patients are selected for a combined heart-kidney transplant if one or more of the following apply:
- End-stage heart and kidney disease not amenable to any other form of therapy
- End-stage heart disease and estimated glomerular filtration rate, or eGFR, is 33 mL/minute or less, or preoperative evaluation of the kidney indicates the likelihood that the rate of progression of renal injury or dysfunction after single organ transplant is high
- End-stage heart disease not amenable to any other form of therapy and associated with a life expectancy of 6 to 12 months
- The consideration for risk-reducing procedure (e.g., CABG) performed at the same time as the organ transplant is a consideration based on medical consultation review
Exclusions:
- Significant systemic or multisystemic disease (other than cardiorenal failure)
- Pulmonary hypertension that is fixed as evidenced by pulmonary vascular resistance, or PVR, greater than 5 Woods units, or trans-pulmonary gradient, or TPG, greater than or equal to 16 mm/Hg.
- Severe pulmonary disease despite optimal medical therapy, not expected to improve with heart transplantation alone.
Potential contraindications for transplant/retransplant:
Note: Final patient eligibility for transplant is subject to the judgment and discretion of the requesting transplant center.
Potential contraindications represent situations where proceeding with transplant isn’t advisable in the context of limited organ availability. Contraindications may evolve over time as transplant experience grows in the medical community. Clinical documentation supplied to the health plan should demonstrate that attending staff at the transplant center have considered all contraindications as part of their overall evaluation of potential organ transplant recipients and have decided to proceed.
- Known current malignancy or history of recent malignancy
- Untreated systemic infection making immunosuppression unsafe, including chronic infection
- Other irreversible end-stage disease not attributed to heart or kidney disease
- Systemic disease that could be exacerbated by immunosuppression
- Psychosocial conditions or chemical dependency affecting ability to adhere to therapy as defined by the transplant program
All transplants must be prior authorized through the Human Organ Transplant Program.
Note: There are individual heart and kidney transplant policies that contain more detailed information. |
38204, 38205, 38206, 38207, 38208,
38209, 38210, 38211, 38212, 38213,
38214, 38215, 38230, 38232, 38240,
38241, 38242, S2140, S2142, S2150
Experimental:
0337U |
Basic benefit and medical policy
BMT HCT for plasma cell dyscrasias
The safety and efficacy of specified bone marrow/hematopoietic cell transplants for plasma cell dyscrasias, including multiple myeloma, plasma cell leukemia, plasmacytoma and POEMS syndrome, have been established. They may be considered useful therapeutic options for patients meeting patient selection criteria.
Inclusionary criteria have been updated, effective Jan. 1, 2024.
Multiple myeloma
Inclusions:
The following hematopoietic cell transplantations for multiple myeloma, including plasma cell leukemia and plasmacytoma, are considered established:
- Single or second salvage (salvage refers to treatments used after a condition hasn’t responded to standard therapy) autologous hematopoietic cell transplantation.
- Tandem transplant with or without maintenance therapy can be considered for any of the following:
- All patients who are candidates for hematopoietic cell transplantation.
- Patients who don’t achieve at least a very good partial response after the first autologous hematopoietic cell transplantation. (A very good partial response, as defined by the International Myeloma Working Group, is a serum and urine M-protein detectable by immunofixation but not on electrophoresis or ≥90% reduction in serum M-protein plus urine M-protein level < 100 mg per 24 hr. Revised based on the new criteria by IMWG).
- Patients with high-risk features.
- Tandem transplantation with an initial round of autologous hematopoietic cell transplantation followed by a non-marrow-ablative conditioning regimen and allogeneic hematopoietic cell transplantation for the treatment of newly diagnosed multiple myeloma patients.
- Myeloablative or nonmyeloablative allogeneic hematopoietic cell transplant is an acceptable option in patients with responsive or primary progressive disease as salvage therapy when these patients have undergone a prior autologous hematopoietic cell transplant.
- Allogeneic HCT should be considered appropriate therapy for any eligible patient with early relapse (less than 24 months) after primary therapy that included an autologous HCT or with high-risk features (i.e., cytogenetics, extramedullary disease, plasma cell leukemia, or high lactate dehydrogenase) provided that they responded favorably to salvage therapy before allogeneic HCT.
Exclusions:
- Allogeneic hematopoietic cell transplantation, myeloablative or nonmyeloablative, as initial therapy of newly diagnosed multiple myeloma is considered experimental.
- More than two tandem transplants, two single transplants or a single and a tandem transplant per patient for the same condition.
- The routine harvesting or storage of an individual’s umbilical cord blood for possible use at some unspecified time in the future.
POEMS syndrome
Inclusions:
Autologous hematopoietic cell transplantation to treat disseminated POEMS syndrome.
Exclusions:
Allogeneic and tandem hematopoietic cell transplantation to treat POEMS syndrome. |
47133, 47135, 47140, 47141, 47142, 47143, 47144, 47145, 47146, 47147, 32850, 32853, 32854, 32855, 32856 |
Basic benefit and medical policy
Lung/double-lung and liver transplant
Combined double-lung and liver transplants have been established as clinically safe and effective for carefully selected individuals with end-stage lung and liver disease when transplantation of a single organ is precluded by severe disease in the other organ system, such that the individual’s prognosis after combined transplantation is felt to be better than with sequential transplantation.
The inclusionary and exclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
Indications for combined lung-double lung and liver transplant include, but aren’t limited to, progressive chronic lung and liver disease unresponsive to other medical and surgical therapy. In general, patients are selected for combined lung-liver transplant if one or more of the following apply:
- A lung transplant is typically required for irreversible, chronic lung diseases for which there is no further medical or surgical therapy available and survival is limited.
- Bilateral lung transplantation is typically required when chronic lung infection disease is present, e.g., associated with cystic fibrosis and bronchiectasis. Some but not all cases of pulmonary hypertension will require bilateral lung transplantation.
- A liver transplant is typically required for irreversibly damaged livers for which there is no further medical or surgical therapy available, prognosis is poor and end stage liver disease (e.g., alcoholic liver disease, viral hepatitis, autoimmune hepatitis, protoporphyria, biliary cirrhosis, vascular disease, trauma or toxic reactions, etc.).
- End-stage lung disease and end-stage liver disease not amenable to any other form of therapy.
The consideration for risk-reducing procedure (e.g., CABG) performed at the same time as the organ transplant is a consideration based on the medical consultation review.
Exclusions:
- Patients with coronary artery disease not amenable to percutaneous intervention or bypass grafting or associated with significant impairment of left ventricular function.
- Patients colonized with highly resistant or highly virulent bacteria, fungi or mycobacteria.
- Patients with intrahepatic cholangiocarcinoma.
- Patients with hepatocellular carcinoma that has extended beyond the liver.
- Patients with ongoing alcohol or drug abuse. (Evidence for abstinence may vary among liver transplant programs but, generally, a minimum of three months is required or enrollment in a sanctioned program.)
Potential contraindications for transplant/retransplant:
Note: Final patient eligibility for transplant is subject to the judgment and discretion of the requesting transplant center.
Potential contraindications represent situations where proceeding with transplant isn’t advisable in the context of limited organ availability. Contraindications may evolve over time as transplant experience grows in the medical community. Clinical documentation supplied to the health plan should demonstrate that attending staff at the transplant center have considered all contraindications as part of their overall evaluation of potential organ transplant recipients and have decided to proceed.
- Known current malignancy or history of recent malignancy
- Untreated systemic infection making immunosuppression unsafe, including chronic infection
- Other irreversible end-stage disease not attributed to liver or lung disease
- Systemic disease that could be exacerbated by immunosuppression
- Psychosocial conditions or chemical dependency affecting ability to adhere to therapy as defined by the transplant program
Liver specific guidelines for alcohol-related hepatitis
- Patients who are being considered for approval for a liver transplant who have liver disease related to alcohol use disorder must be evaluated for ongoing alcohol use.
- To determine candidacy for liver transplant in the setting of alcohol related hepatitis, guidelines such as the Dallas consensus criteria and the SALT criteria must be met.
All transplants must be prior authorized through the Human Organ Transplant Program.
Note: There are individual transplant policies for each of these organs (Lung Transplant, Liver Transplant) that contain more detailed information. |
47135, 47399,** 50360, 50365
**Unlisted procedure
|
Basic benefit and medical policy
Transplant of liver and kidney (combined)
Combined liver-kidney transplants have been clinically established as safe and effective for carefully selected individuals with end-stage disease in both organs.
The inclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
Indications for combined liver-kidney transplant include, but aren’t limited to, progressive chronic kidney/liver disease unresponsive to other medical and surgical therapy. In general, individuals are selected for combined kidney-liver transplant if one or more of the following apply:
- End-stage kidney and liver disease not amenable to any other form of therapy
- End-stage liver disease and estimated glomerular filtration rate, or eGFR, is 33 mL/minute or less, or preoperative evaluation of the kidney indicates the likelihood that the rate of progression of renal injury or dysfunction after single organ transplant is high
- Polycystic kidney and liver disease, primary hyperoxaluria and hepatorenal syndrome with terminal implications
- Fulminant or sub-acute hepatic/kidney failure
The consideration for risk-reducing procedure (e.g., CABG) performed at the same time as the organ transplant is a consideration based on the medical consultation review.
Note: Final patient eligibility for combined liver-kidney transplant is subject to the judgment and discretion of the requesting transplant center. Refer to the Liver Transplant policy for full inclusionary criteria for liver transplant patients, and Kidney Transplant policy for full inclusionary criteria for kidney transplant patients.
Exclusions:
- Significant systemic or multisystemic disease (other than hepatorenal failure)
- Patients with ongoing alcohol or drug abuse. (Evidence for abstinence may vary among liver transplant programs, but generally, a minimum of three months is required or enrollment in a sanctioned program.)
- Malignancies metastasized to or extending beyond the margins of the liver or kidney.
- Individuals not meeting full inclusionary guidelines for liver transplant or renal transplant alone.
Potential contraindications for transplant/retransplant:
Note: Final patient eligibility for transplant is subject to the judgment and discretion of the requesting transplant center.
Potential contraindications represent situations where proceeding with the transplant isn’t advisable in the context of limited organ availability. Contraindications may evolve over time as transplant experience grows in the medical community. Clinical documentation supplied to the health plan should demonstrate that attending staff at the transplant center have considered all contraindications as part of their overall evaluation of potential organ transplant recipients and have decided to proceed.
- Known current malignancy, or history of recent malignancy
- Untreated systemic infection making immunosuppression unsafe, including chronic infection
- Other irreversible end-stage disease not attributed to liver or kidney disease
- Systemic disease that could be exacerbated by immunosuppression
- Psychosocial conditions or chemical dependency affecting ability to adhere to therapy as defined by the transplant program
Liver-specific guidelines for alcohol-related hepatitis:
- Patients who are being considered for approval for a liver transplant who have liver disease related to alcohol use disorder must be evaluated for ongoing alcohol use.
- To determine candidacy for liver transplant in the setting of alcohol related hepatitis, guidelines such as the Dallas consensus criteria and the SALT criteria must be met.
All transplants must be prior authorized through the Human Organ Transplant Program
Note: There are individual policies for each of these organs (liver transplant, kidney transplant) that contain more detailed information. |
66179, 66180, 66183, 66184, 66185,
0449T, 0450T, 0474T, 0671T, 66999
Note: The above procedures may be billed with cataract removal codes 66982-66984, 66987-66989, 66991
Experimental/not covered:
66999,** 0253T
**Not otherwise classified procedure |
Basic benefit and medical policy Aqueous shunts and stents for glaucoma
The safety and effectiveness of the insertion of U.S. Food and Drug Administration-approved aqueous shunts and stents have been established. They are useful therapeutic options for reducing intraocular pressure in individuals with glaucoma in whom medical therapy has failed to adequately control intraocular pressure.
Insertion of ab externo aqueous shunts approved by the FDA is established as a method to reduce intraocular pressure in individuals with glaucoma in whom medical therapy has failed to adequately control intraocular pressure.
Use of an ab externo aqueous shunt for all other conditions, including in individuals with glaucoma when intraocular pressure is adequately controlled by medications, is considered experimental.
Insertion of ab interno aqueous stents approved by the FDA as a method to reduce intraocular pressure in individuals with glaucoma in whom medical therapy has failed to adequately control intraocular pressure, is considered established.
Implantation of one or two FDA-approved microstents in conjunction with cataract surgery may be considered established in individuals with mild to moderate open-angle glaucoma currently treated with ocular hypotensive medication.
The use of ab interno stents for all other conditions is considered experimental.
Inclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
Insertion of FDA-approved aqueous shunts is considered established as a method to reduce intraocular pressure in patients with mild to moderate open-angle glaucoma when conventional pharmacologic treatments have failed to control intraocular pressure adequately.
Currently available FDA-approved shunts include:
- Ahmed™ glaucoma implant
- Baerveldt® seton
- Ex-PRESS® mini glaucoma shunt
- Glaucoma pressure regulator
- Krupin-Denver valve implant
- Molteno® implant
- Schocket shunt
- Xen Gel Stent
- CyPass® Micro-Stenta (recalled)
- iStent® a
- iStent Inject® ab
- iStent Infinate®
- Hydrus™ ®
aThese stents are indicated for use in conjunction with cataract surgery for the reduction of IOP in adult patients with mild to moderate primary open-angle glaucoma.
abThe iStent Inject® comes pre-loaded with two stents.
Exclusions:
- The use of an aqueous shunt for all other conditions, including patients with glaucoma when intraocular pressure, is controlled by medications.
- Insertion of aqueous shunts that aren’t FDA approved.
- For the Trabecular Micro-Bypass iStent and the iStent Inject, patients with the following conditions aren’t appropriate candidates and the insertion of this stent would be considered experimental:
- Quick or sudden increase in eye pressure
- Inflammation of the eye tissue (uvea)
- Neovascular glaucoma
- Noticeable birth irregularities on the front of the eye
- Orbital tumor
- Thyroid eye disease
- Sturge-Weber syndrome
- Any other type of condition that may cause elevated pressure in the veins of the eye
- For the Hydrus Microstent, patients with the following conditions aren’t appropriate candidates and the insertion of this stent would be considered experimental:
- When the colored part of the eye (iris) is pushed up against the drainage pathway or when other material blocks the drainage pathway
- Traumatic glaucoma, malignant glaucoma or inflammation of the eye tissue
- Glaucoma associated with the growth of abnormal blood vessels in the eye
- Noticeable birth irregularities of the anterior chamber angle
|
75574, 75580 |
Basic benefit and medical policy
Coronary CT angiography with selected noninvasive FFR
The use of noninvasive fractional flow reserve to guide decisions about the use of invasive coronary angiography in select individuals has been established. It’s a useful diagnostic option when indicated.
Inclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
Individuals must meet both criteria:
- Have stable chest pain
- Have intermediate risk of coronary artery disease (i.e., suspected or presumed stable ischemic heart disease)
And
Meet one of the following:
- Diagnosis of congestive heart failure/left ventricular dysfunction when all the following are met:
- Left ventricular ejection fraction < 55%
- Low to moderate coronary heart disease riska
- Symptomaticb or asymptomatic individuals undergoing non-coronary surgery (including open and percutaneous valvular procedures or ascending aortic surgery)
- All the pre-operative information can be obtained using cardiac CT
And
- Moderate coronary heart disease riska
- Symptomaticb individuals who are suspected of having coronary artery disease and meet one of the following:
- During a planned outpatient exercise stress test (without imaging), all the following apply:
- Performed within the past 60 days
- Individual is symptomaticb
- During the test one of the following occurred:
- Exercise-induced chest pain
- ST segment change
- Abnormal blood pressure response
- Complex ventricular arrhythmias
- Have undergone either myocardial perfusion imaging or a stress echocardiogram within the past 60 days and imaging is one of the following:
- Neither normal or abnormal
- Abnormal
- No coronary artery disease imaging has been performed within the preceding 60 days (e.g., Myocardial perfusion imaging, cardiac PET scan, stress echo or coronary angiogram)
- Symptomaticb individual with abnormal resting EKG
- Exercise stress test (without imaging) would be uninterpretable related to one of the following:
- Left bundle branch block
- Paced ventricular rhythm
- Left ventricular hypertrophy with repolarization abnormalities
- Resting ST segment depression
- Digoxin effects as evidence by one of the following:
- ST depression in a concave shape
- Flattened, inverted, or biphasic T waves
- Shortened QT interval
- Pre-excitation syndrome (i.e. Lown-Ganong-Levine Syndrome, Wolff-Parkinson-White Syndrome)
- Short PR interval (< 0.12 sec)
Note: Fractional flow reserve using coronary tomography angiography requires at least 64-slice coronary computed tomography angiography and cannot be calculated when images lack sufficient quality
aRisk factor is determined using standard assessment methods (i.e., SCORE risk chart)
bSymptomatic is defined by one or more of the following:
- Chest pain with low probability of coronary artery disease, but high risk
- Moderate to high risk of coronary artery disease and one of the following:
- Chest, jaw, neck, shoulder, arm, hand, epigastric or back pain
- Diaphoresis
- Syncope
- Shortness of breath
- High risk of coronary artery disease and one of the following:
- Palpitations
- Lightheadedness
- Near syncope
- Nausea/vomiting
- Anxiety
- Weakness
- Fatigue
- Individuals with any cardiac symptom who have any of the following diseases associated with coronary artery disease
- Abdominal aortic aneurysm
- Chronic renal insufficiency or renal failure
- Diabetes mellitus
- Established and symptomatic peripheral vascular disease
- History of:
- Cerebrovascular accident
- Transient ischemic attack
- Carotid endarterectomy
- High grade carotid stenosis (>70%)
Exclusions:
- Assessment of coronary arteries for suspected congenital anomalies
- Individuals who have:
- BMI > 35% kg/m2
- Presence of uncontrolled rapid heart rate or arrhythmia
- Suspicion of acute coronary syndrome when acute myocardial infarction or unstable angina have not been ruled out
- History of:
- Myocardial infarction within the last 30 days
- Coronary artery bypass graft surgery
- Presence of dense arterial calcification or intracoronary stent
- Evidence of clinical instability (e.g., unstable blood pressure – Systolic < 90 mmHg, severe congestive heart failure, acute pulmonary edema, cardiogenic shock)
- Individuals who require emergent procedures
- Individuals not meeting inclusionary guidelines
|
81210, 81445, 81455, 81456, 0037U, 0239U, 0239U, 0242U, 0326U |
Basic benefit and medical policy
GT – BRAF mutation in melanoma targeted therapy
Testing for BRAF V600 variants in tumor tissue of individuals with unresectable or metastatic melanoma or with resected stage III melanoma is established to select individuals for treatment with FDA-approved BRAF inhibitors, MEK inhibitors or immunotherapy.
Testing for BRAF V600 variants for all other individuals with melanoma is considered experimental.
Testing for genetic mutations using a panel with 5-50 genes may also be considered appropriate for cutaneous melanoma (stage 3 and stage 4).
Testing for genetic mutations using a panel with 51 or more genes may also be considered appropriate for cutaneous melanoma (stage 3 and stage 4).
Molecular testing on peripheral blood (i.e., liquid biopsy, ctDNA) may be established when criteria are met.
The medical policy statement and inclusionary and exclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusionary and exclusionary guidelines:
Inclusions:
Refer to the pharmacy Genetic Testing of Drugs policy (Atezolizumab, Binimetinib, Dabrafenib, Encorafenib, Entrectinib, Larotrectinib, Pembrolizumab, Vemurafenib, Trametinib and Cobimetinib) for patient selection.
Circulating tumor DNA (liquid biopsy)
The clinical utility of circulating tumor DNA and circulating tumor cells for management of advanced solid cancers has been established when all the following criteria is met.
- May be considered established for guidance in the selection of appropriate targeted FDA therapeutic options for any of the following conditions:
- Metastatic cancers
- Inoperable locally advanced cancers
- Refractory cancers
- Recurrent cancers
- Advanced cancer (stages 3 or 4)
- Individual hasn’t been previously tested using the same liquid biopsy panel, unless a new primary cancer diagnosis is made, and further cancer treatment is being considered or the individual is experiencing a relapse.
- There is clinical documentation that tissue-based testing can’t be performed (e.g., insufficient sample, inaccessible tumor or where there may be a delay in obtaining tumor sample) or tissue-based testing isn’t required when there is an FDA-approved companion diagnostic device that is a circulating tumor test (liquid biopsy panel).
FDA-approved companion diagnostics
A companion diagnostic is an FDA-approved medical device, often an in vitro device, which provides information that is essential for the safe and effective use of a corresponding drug or biological product. The test helps a health care professional determine whether, for a specific patient, a particular therapeutic product’s benefits outweigh any potential serious side effects or risks.
Companion diagnostics can:
- Identify patients who are most likely to benefit from a particular therapeutic product.
- Identify patients likely to be at increased risk for serious side effects as a result of treatment with a particular therapeutic product.
- Monitor response to treatment with a particular therapeutic product for the purpose of adjusting treatment to achieve improved safety or effectiveness.
FDA-approved companion diagnostic tests
FDA-approved companion diagnostic tests include:
- Tests that are billed with CPT codes (most laboratories are able to process these)
- Proprietary laboratory analyses, or PLA, tests (processed by one specific independent laboratory).
Most PLA tests have billing codes that end in “U.”
Proprietary laboratory analyses testing
A PLA test is considered established when the following criteria is met:
- Biomarker confirmation is required by an FDA-approved or cleared test before initiating treatment (as described in the FDA prescribing label of the therapeutic in the section “Indications and Usage”).
- The test is an FDA-approved companion diagnostic.
Information regarding FDA-approved companion diagnostic tests should be obtained from the FDA’s List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools) webpage.
For accuracy, please access the information directly from the FDA site as the website is updated frequently.
Exclusions:
- The use of circulating tumor DNA and circulating tumor cells is considered experimental when the criteria above aren’t met.
- The use of circulating tumor DNA and circulating tumor cell testing is considered experimental for all other indications related to solid tumors, including measurable residual disease, or MRD, testing and cancer screening (e.g., Galleri).
|
81351, 81352, 81353, 81432
Experimental/not covered:
0102U, 0131U, 81479 |
Basic benefit and medical policy
Genetic testing for Li-Fraumeni syndrome
The safety and effectiveness of genetic testing for TP53 to confirm a diagnosis of Li-Fraumeni syndrome and pediatric hypoliploid acute lymphoblastic leukemia have been established. Genetic testing may be considered a useful diagnostic tool when indicated and should be performed in conjunction with appropriate pre-and post-test genetic counseling.
Inclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
To confirm a diagnosis of Li-Fraumeni syndrome under the following conditions:
- In an individual who meets either the classic or the Chompret clinical diagnostic criteria for Li-Fraumeni syndrome
- In individuals with early-onset breast cancer (age of diagnosis <31 years)
- Pediatric hypodiploid acute lymphoblastic leukemia**
- For carrier or presymptomatic testing in relatives of individuals with known TP53 gene variants.
**The NCCN Pediatric Acute Lymphoblastic Leukemia panel considers “pediatric” to include any patient age ≤18 years, as well as adolescent and young adult patients >18 years treated in a pediatric oncology setting; the latter could include patients up to age 30 years.
Exclusions:
Genetic testing for a germline TP53 variant for all other indications.
Chompret criteria
Chompret, et al. (2001), developed criteria that has the highest positive predictive value and that, when combined with the classic LFS criteria, provides the highest sensitivity for identifying individuals with LFS. The Chompret criteria was updated in 2009 to assist in identifying families with milder phenotypes. The Chompret criteria will also identify individuals with de novo TP53 pathogenic variants, whereas the classic LFS criteria require a family history.
The Chompret criteria, most recently updated in 2015, is defined as the following:
- Proband with tumor belonging to the LFS tumor spectrum (e.g., soft tissue sarcoma, osteosarcoma, CNS tumor, premenopausal breast cancer, adrenocortical carcinoma) before age 46 years and at least one, first- or second-degree relative with LFS tumor (except breast cancer if the proband has breast cancer) before age 56 or with multiple tumors
- Proband with multiple tumors (except multiple breast tumors), two of which belong to the LFS tumor spectrum and the first of which occurred before age 46
- Patient with adrenocortical carcinoma, rhabdomyosarcoma of embryonal anaplastic subtype, or choroid plexus tumor, irrespective of family history
- Proband with breast cancer before age 31
Testing criteria for Li-Fraumeni syndrome
Testing is clinically indicated in the following scenarios:
general testing criteria:
- Individual from a family with a known TP53 P/LP variant
- Classic Li-Fraumeni syndrome criteria:
- Combination of an individual diagnosed at age <45 years with a sarcoma and a first-degree relative diagnosed at age <45 years with cancer and an additional first- or second-degree relative in the same lineage with cancer diagnosed at age <45 years, or a sarcoma at any age
Chompret criteria (one of the following):
- Individual with a tumor from LFS tumor spectrum (e.g., soft tissue sarcoma, osteosarcoma, central nervous system tumor, breast cancer, adrenocortical carcinoma), before 46 years of age and at least one first- or second-degree relative with any of the aforementioned cancers (other than breast cancer if the proband has breast cancer) before the age of 56 years or with multiple primaries at any age.
- Individual with multiple tumors (except multiple breast tumors), two of which belong to LFS tumor spectrum with the initial cancer occurring before the age of 46 years.
- Individual with ACC, or choroid plexus carcinoma or rhabdomyosarcoma of embryonal anaplastic subtype, at any age of onset, regardless of family history.
- Breast cancer before 31 years of age.
- Personal or family history of pediatric hypodiploid acute lymphoblastic leukemia.
- In individuals with cancer with a P/LP TP53 variant identified on tumor-only genomic testing, germline testing should be considered for:
- Those meeting one or more of the other LFS testing criterion above after reevaluation of personal and family history
- Those diagnosed <30 years of age with any cancer
- Those with clinical scenario not meeting these criteria but warranting germline evaluation per clinical discretion
National Comprehensive Cancer Network guidelines recommend TP53 testing for individuals who meet classic LFS criteria and Chompret criteria.
|
81445, 81450, 81455, 0239U, 0242U, 0326U
Experimental
86152, 86153, 0091U, 0338U |
Basic benefit and medical policy
Circulating tumor DNA (liquid biopsy)
The clinical utility of circulating tumor DNA and circulating tumor cells for selecting targeted therapy for advanced solid cancers has been established when criteria is met.
The use of circulating tumor DNA and circulating tumor cell testing is considered experimental for all other indications related to solid tumors, including measurable residual disease, or MRD, testing and cancer screening (e.g., Galleri).
The medical policy statement, and inclusionary and exclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusionary and exclusionary guidelines:
Inclusions:
The clinical utility of circulating tumor DNA and circulating tumor cells for selecting targeted therapy for advanced solid cancers has been established when all the following criteria is met.
- May be considered established for guidance in the selection of appropriate targeted FDA therapeutic options for any of the following conditions:
- Metastatic cancers
- Inoperable locally advanced cancers
- Refractory cancers
- Recurrent cancers
- Advanced cancer (stages III or IV)
- Individual hasn’t been previously tested using the same liquid biopsy panel, unless a new primary cancer diagnosis is made, and further cancer treatment is being considered or the individual is experiencing a relapse.
- There is clinical documentation that tissue-based testing can’t be performed (e.g., insufficient sample, inaccessible tumor or where there may be a delay in obtaining tumor sample) or tissue-based testing isn’t required when there is an FDA-approved companion diagnostic device that is a circulating tumor test (liquid biopsy).
U.S. Food and Drug Administration companion diagnostics
A companion diagnostic is an FDA-approved medical device, often an in vitro device, which provides information that is essential for the safe and effective use of a corresponding drug or biological product. The test helps a health care professional determine whether, for a specific patient, a particular therapeutic product’s benefits outweigh any potential serious side effects or risks.
Companion diagnostics can:
- Identify patients who are most likely to benefit from a particular therapeutic product.
- Identify patients likely to be at increased risk for serious side effects as a result of treatment with a particular therapeutic product.
- Monitor response to treatment with a particular therapeutic product for the purpose of adjusting treatment to achieve improved safety or effectiveness.
FDA-approved companion diagnostic tests
FDA-approved companion diagnostic tests include:
- Tests billed with CPT codes (most laboratories are able to process these)
- Proprietary laboratory analyses, or PLA tests (processed by one specific independent laboratory). Most PLA tests have billing codes that end in “U.”
Proprietary laboratory analyses testing
A PLA test is considered established when the following criteria is met:
- Biomarker confirmation is required by an FDA-approved or cleared test prior to initiating treatment (as described in the FDA prescribing label of the therapeutic in the section “Indications and Usage”).
- The test is an FDA-approved companion diagnostic.
Information regarding FDA-approved companion diagnostic tests should be obtained from the FDA’s List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools) webpage.
For accuracy, access the information directly from the FDA site, which is updated frequently.
Exclusions:
- The use of circulating tumor DNA and circulating tumor cells is considered experimental when criteria above aren’t met.
- The use of circulating tumor DNA and circulating tumor cell testing is considered experimental for all other indications related to solid tumors, including measurable residual disease, or MRD, testing and cancer screening (e.g., Galleri).
|
83993 |
Basic benefit and medical policy
Fecal calprotectin
The clinical utility of fecal calprotectin testing has been established for adult and pediatric individuals. It can be a useful option when criteria has been met.
The medical policy statement and inclusionary and exclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusionary and exclusionary guidelines:
Inclusions:
Children
- As an adjunctive non-invasive test to confirm diagnosis of inflammatory bowel disease, or IBD
- For confirming a recurrence or relapse of IBD
- To determine if endoscopy may be needed
Adults
- To differentiate between inflammatory bowel disease and non-inflammatory bowel disease (including irritable bowel syndrome)
- For monitoring of gastrointestinal conditions such as inflammatory bowel disease
- To assess response to therapy and relapse for inflammatory bowel disease
Exclusions:
- The use of fecal calprotectin testing in any other clinical situation
|
97039,** 64999**
**Not otherwise classified codes |
Basic benefit and medical policy
PENS, PNT and restorative neurostimulation therapy
Percutaneous electrical nerve stimulation, or PENS, percutaneous neuromodulation therapy, known as PNT, and restorative neurostimulation therapy are considered experimental for chronic pain. The effectiveness of these therapies hasn’t been clinically proven. This policy statement has been updated, effective Jan. 1, 2024. |
Condition codes 35 and 92 |
Basic benefit and medical policy
Condition codes added
The National Uniform Billing Committee added condition codes 35 and 92, effective Jan. 1, 2024. |
C9399
J3490
J3590 |
Basic benefit and medical policy
Prevymis (letermovir)
The FDA updated the indications for Prevymis (letermovir). This was effective June 5, 2023.
- Prophylaxis of CMV disease in adult kidney transplant recipients at high risk (donor CMV seropositive/recipient CMV seronegative [D+/R-]).
Dosage and administration:
Kidney transplant: 480 mg administered once daily orally or as an IV infusion over one hour through 200 days post-transplant. |
C9769, 53855 |
Basic benefit and medical policy
Prostatic stents and implants for BPH
The use of a temporarily implanted nitinol device (e.g., iTind) for treatment of lower urinary tract symptoms due to benign prostatic hyperplasia is considered experimental. The evidence is insufficient to determine that the technology results in an improvement in the net health outcome.
Placement of temporary prostatic stents (e.g., Spanner™) is experimental for all uses, including, but not limited to BPH, following surgical treatment of BPH, prostate cancer or radiation therapy. They haven’t been scientifically demonstrated to be as safe and effective as conventional treatment and haven’t been shown to improve net health outcomes.
This policy update is effective Jan. 1, 2024. |
E0650, E0651, E0652, E0655, E0656, E0657, E0660, E0665, E0666, E0667, E0668, E0669, E0670, E0671, E0672, E0673, E0676 |
Basic benefit and medical policy
Postsurgical home use of limb compression devices
The safety and effectiveness of postsurgical home limb compression devices for venous thromboembolism prophylaxis have been established. It may be considered a useful therapeutic option when clinical criteria are met.
Inclusionary and exclusionary criteria have been updated, effective Jan. 1, 2024.
Inclusions:
Postsurgical home use of limb compression devices for venous thromboembolism, or VTE, prophylaxis is indicated when one of the following criteria are met:
- After major orthopedic proceduresa in individuals with a contraindicationb to anticoagulant and antiplatelet agents (e.g., at high-risk for bleeding)
- After major non-orthopedic or other orthopedic procedures in individuals who are at moderate or high risk of venous thromboembolism (see Tables IE 1 and 2) with a contraindicationb to anticoagulant and antiplatelet agents (e.g., at high-risk for bleeding)
- After major orthopedica or major non-orthopedic proceduresc, as an adjunct to anticoagulant or antiplatelet therapy, in individuals who are at extremely high riskd for venous thromboembolism.
Exclusions:
- Individuals who are at low risk of venous thromboembolism
- Home use of limb compression devices for venous thromboembolism prophylaxis for periods longer than 30 days post-surgery
aExamples include major orthopedic surgery includes total hip arthroplasty, total knee arthroplasty or hip fracture surgery.
bThe main contraindication to anticoagulants is a high risk of bleeding. However, there is no absolute threshold at which anticoagulants cannot be used. Rather, there is a risk-benefit continuum that takes into account the benefits of treatment and risks of bleeding. There may also be intolerance to specific agents, although uncommon. Intolerance may result from allergic reactions or adverse events. Finally, when heparin preparations are used, serum antibodies and heparin-induced thrombocytopenia can develop, precluding further use of heparin products.
cExamples of major non-orthopedic surgery may include urological, abdominal, pelvic, neurological and extensive trauma procedures.
dIndividuals older than 60 years plus prior VTE, cancer or molecular hypercoagulable state.

|
J0174 |
Basic benefit and medical policy
Leqembi (Lecanemab-irmbis)
Effective July 6, 2023, documentation was modified to reflect that the experimental drug Leqembi (Lecanemab-irmbis) is no longer indicated in the drug’s description for the following usage:
- There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in amyloid beta plaques observed in patients treated with Leqembi (Lecanemab-irmb. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
This applies to groups that have opted into the Medical Drug Prior Authorization Program.
|
J3490
J3590 |
Basic benefit and medical policy
Rystiggo (rozanolixizumab-noli)
Rystiggo (rozanolixizumab-noli) is considered established when criteria are met, effective June 27, 2023.
Coverage of Rystiggo (rozanolixizumab-noli) is provided when all the following are met:
- FDA-approved indication
- FDA-approved age
- Documented anti-acetylcholine receptor, or AChR, antibody positive myasthenia gravis, or MG, identified by both of the following:
- Lab record or chart notes identifying the patient is positive for anti-AChR antibodies
- One of the following confirmatory tests:
- Positive edrophonium test
- History of clinical response to oral cholinesterase inhibitors (for example, pyridostigmine)
- Electrophysiological evidence of abnormal neuromuscular transmission by repetitive nerve stimulation, or RNS, or single-fiber electromyography, or SFEMG
Or
- Documented anti-muscle-specific tyrosine kinase, or MuSK, antibody positive MG identified by both of the following:
- Lab record or chart notes identifying the patient is positive for anti-MuSK antibodies.
- Electrophysiological evidence of abnormal neuromuscular transmission by repetitive nerve stimulation, or RNS, or single-fiber electromyography, or SFEMG.
- Patients must not have a history of:
- Thymectomy within six months
- Current thymoma
- Other neoplasms of the thymus
- Previous treatment courses of at least 12 weeks with one of the following standards of care have been ineffective: methotrexate, azathioprine, cyclophosphamide, cyclosporine, mycophenolate mofetil, or tacrolimus unless all are contraindicated or not tolerated.
- Patient is currently receiving, and will continue to receive, a stable standard of care regimen.
- Must not be used with other biologic therapies for myasthenia gravis or immunoglobulin therapy.
- Trial and failure, intolerance or a contraindication to the preferred products as specified in the Blue Cross Blue Shield of Michigan and Blue Care Network medical utilization management drug list.
Quantity limitations, authorization period and renewal criteria:
- Quantity limits: Align with FDA-recommended dosing.
- Authorization period: One year at a time
- Renewal criteria: Clinical documentation must be provided to confirm that current criteria are met and that the medication is providing clinical benefit.
Rystiggo (rozanolixizumab-noli) isn’t a benefit for URMBT.
|
J3490
J3590 |
Basic benefit and medical policy
Talvey (talquetamab-tgvs)
Talvey (talquetamab-tgvs) is considered established when criteria are met, effective Aug. 10, 2023.
Talvey is a bispecific GPRC5D-directed CD3 T-cell engager indicated for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least four prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody.
This indication is approved under accelerated approval based on response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
Dosage and administration:
- For subcutaneous injection.
- Patients should be hospitalized for 48 hours after all doses within the step-up dosing schedule.
- Administer pretreatment medications as recommended.
Talvey weekly dosing schedule
Dosing schedule
Step-up dosing schedule
Day 1
Step-up dose 1
Dose: 0.01 mg/kg
Day 4
Step-up dose 2
Dose: 0.06 mg/kg
Day 7
First treatment dose
Dose: 0.4 mg/kg once weekly
Weekly dosing schedule
One week after the first treatment dose and weekly thereafter
Subsequent treatment doses
Dose: 0.4 mg/kg once weekly
Talvey biweekly dosing schedule
Dosing schedule
Step-up dosing schedule
Day 1
Step-up dose 1
Dose: 0.01 mg/kg
Day 4
Step-up dose 2
Dose: 0.06 mg/kg
Day 7
Step-up dose 3
Dose: 0.4 mg/kg once weekly
Day 10
First treatment dose
Dose: 0.8 mg/kg once weekly
Biweekly (every two weeks) dosing schedule
Two weeks after the first treatment dose and every week thereafter
Subsequent treatment doses
Dose: 0.8 mg/kg every 2 weeks
Dosage forms and strengths:
Injection
- 3 mg/1.5 mL (2 mg/mL) in a single-dose vial
- 40 mg/mL in a single-dose vial
Talvey (talquetamab-tgvs) isn’t a benefit for URMBT. |
J3490
J3590 |
Basic benefit and medical policy
Tyruko (natalizumab-sztn)
Effective Aug. 24, 2023, Tyruko (natalizumab-sztn) is covered for its FDA-approved indications.
Coverage of Tyruko (natalizumab-sztn) is provided when all the following are met:
- Diagnosis of multiple sclerosis:
- FDA-approved age
- FDA-approved indication
- Won’t be used in combination with other disease-modifying treatments of multiple sclerosis
- Diagnosis of Crohn’s disease
- Coverage will be provided for biosimilar products for FDA-labeled indications of the innovator product when criteria are met.
- Trial and failure, contraindication or intolerance to the preferred drugs as listed in Blue Cross Blue Shield of Michigan and Blue Care Network’s utilization management medical drug list.
Quantity limitations, authorization period and renewal criteria:
- Quantity limits: Align with FDA-recommended dosing.
- Authorization period: One year at a time
- Renewal criteria: Clinical documentation must be provided to confirm that current criteria are met and that the medication is providing clinical benefit.
This drug isn’t a benefit for URMBT.
|
J3490
J3590 |
Basic benefit and medical policy
Veopoz (pozelimab-bbfg IV/SQ)
Effective Aug. 18, 2023, Veopoz (pozelimab-bbfg IV/SQ) is covered for its FDA-approved indications.
Coverage of Veopoz (pozelimab-bbfg IV/SQ) is provided when all the following are met:
- FDA-approved indication
- FDA-approved age
- Confirmed biallelic CD55 loss-of-function mutation
- Trial and failure, intolerance or a contraindication to Soliris or a Soliris biosimilar
- Must not be used in combination with Soliris or any other C5 complement inhibitor
- Trial and failure, intolerance or a contraindication to the preferred products as specified in the Blue Cross Blue Shield of Michigan or Blue Care Network medical utilization management drug list
Quantity limitations, authorization period and renewal criteria
- Quantity limits: Align with FDA-recommended dosing
- Authorization period: One year at a time
- Renewal criteria: Clinical documentation must be provided to confirm that current criteria are met and that the medication is providing clinical benefit
This drug isn’t a benefit for URMBT.
|
J9217 |
Basic benefit and medical policy
Eligard (leuprolide acetate for depot suspension)
Effective July 20, 2023, Eligard (leuprolide acetate for depot suspension) is covered for the following FDA-approved indications:
Eligard (leuprolide acetate for depot suspension) is a gonadotropin releasing hormone, or GnRH, agonist indicated for the treatment of advanced prostate cancer. |
J9272 |
Basic benefit and medical policy
Jemperli (dostarlimab-gxly)
The FDA has updated the indications for Jemperli (dostarlimab-gxly). This was effective July 31, 2023.
Endometrial cancer
- In combination with carboplatin and paclitaxel, followed by Jemperli (dostarlimab-gxly) as a single agent for the treatment of adult patients with primary advanced or recurrent endometrial cancer that is mismatch repair deficient, or dMMR, as determined by an FDA-approved test, or microsatellite instability-high, known as MSI-H.
- As a single agent for the treatment of adult patients with dMMR recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following prior treatment with a platinum-containing regimen in any setting and aren’t candidates for curative surgery or radiation.
Dosage and administration:
- Jemperli (dostarlimab-gxly), in combination with carboplatin and paclitaxel, for dMMR or MSI-H primary advanced or recurrent endometrial cancer: 500 mg every three weeks for six doses followed by 1,000 mg monotherapy every six weeks.
- Jemperli (dostarlimab-gxly), as a single-agent, for dMMR recurrent or advanced endometrial cancer: 500 mg every three weeks for four doses followed by 1,000 mg every six weeks.
|
J9355
Q5112
Q5113
Q5114
Q5116
Q5117 |
Basic benefit and medical policy
Off-label use approved
Blue Cross Blue Shield of Michigan has approved payment for the off-label use of Herceptin (trastuzumab), Ontruzant (trastuzumab-dttb), Herzuma (trastuzumab-pkrb), Ogivri (trastuzumab-dkst), Trazimera (trastuzumab-qyyp) and Kanjinti (trastuzumab-anns) for the treatment of malignant neoplasm of the endometrium.
URMBT is excluded from this change. |
None of the information included in this billing chart is intended to be legal advice and, as such, it remains the provider’s responsibility to ensure that all coding and documentation are done in accordance with all applicable state and federal laws and regulations. |
